17-idrossiprogesterone
Noto anche come: 17-OHP; 17-OH progesterone; Progesterone -17-OH
Nome ufficiale: 17-idrossiprogesterone
Ultima Revisione: 19.03.2018.
Ultima Modifica: 03.12.2018
Articolo approvato dal Comitato editoriale di labtestsonline.org ed in corso di revisione da parte del Comitato editoriale italiano
In Sintesi
Perché?
Per la ricerca, diagnosi e monitoraggio del trattamento dell’iperplasia surrenale congenita(sindromesurreno-genitale;CAH); talvolta per escludere altre patologie con sintomi analoghi.
Quando?
E' un esame che,in alcune regioni, viene eseguito all’interno dei protocolli di screening neonatale. Viene inoltre richiesto quando un bambino presenta genitali ambigui, quando una bambina manifesta irsutismo o altri sintomi di virilizzazione, quando un bambino sviluppa prematuramente i caratteri sessuali e per controllare periodicamente il trattamento dellaCAH.
Il campione
Campioni di sangue prelevati da una vena dell’avambraccio o dal tallone di un bambino (per lo screening neonatale).
La preparazione
No, ma può essere richiesto un prelievo effettuato nelle prime ore del giorno; alle donne potrebbe anche essere richiesto di effettuare il prelievo in una specifica fase del ciclo mestruale. Il test non dovrebbe essere richiesto in pazienti in cura con steroidi.
L'Esame
Questo test misura la quantità di 17-idrossiprogesterone (17-OHP) nel sangue. Il 17-OHP è un ormone steroideo, precursore del cortisolo. La misura del 17-OHP è utile nella rilevazione e nel monitoraggio dell’iperplasia surrenale congenita (sindrome surreno-genitale, CAH), una condizione ereditaria che determina la diminuzione di cortisolo e aldosterone e l’aumento della produzione degli ormoni sessuali maschili (androgeni).
Il 17-OHP deriva dal colesterolo. Non si tratta di un ormone nella forma attiva ma di un precursore ormonale.
Il cortisolo è un ormone prodotto dalle ghiandole surrenali. È necessario per la degradazione delle proteine, del glucosio e dei lipidi, mantiene costante la pressione sanguigna e regola il sistema immunitario. Le ghiandole surrenali producono anche altri ormoni steroidei, come l’aldosterone, necessario nella regolazione del sodio e del potassio nel sangue e della pressione sanguigna, e gli androgeni, sostanze che, come il testosterone, tra le altre cose, sono responsabili dello sviluppo dei caratteri sessuali secondari maschili.
Per completare la produzione di cortisolo sono necessari numerosi enzimi. Se uno o più degli enzimi necessari è insufficiente o mal funzionante, i livelli di cortisolo prodotti risultano inadeguati e i precursori del cortisolo, come il 17-OHP, si accumulano nel sangue. Di conseguenza si ha lo sviluppo della CAH.
La causa più frequente (circa il 90% dei casi) della CAH è la perdita totale o parziale dell’enzima 21-idrossilasi.
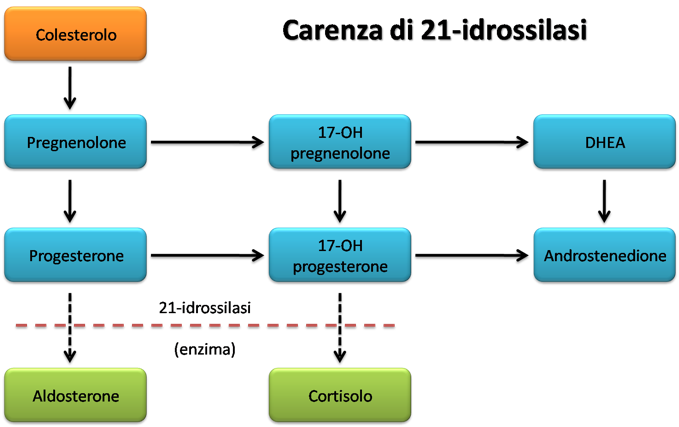
Nelle ghiandole surrenali il colesterolo viene convertito in pregnenolone, un precursore di aldosterone, cortisolo e androgeni. Un deficit di 21-idrossilasi determina la diminuzione di aldosterone e cortisolo (in verde) e l’accumulo dei precursori. Questo deficit enzimatico è ereditario ed è la causa più frequente di CAH.
I bassi livelli di cortisolo stimolano la produzione, ad opera della ghiandola pituitaria (ipofisi), dell'ormone adrenocorticotropo (ACTH), responsabile dell’accrescimento del surrene (iperplasia surrenale). L’aumento delle dimensioni e dell’attività del surrene tuttavia non determina un aumento della produzione di cortisolo; vi è però un accumulo dei precursori, come il 17-OHP. Per questo motivo l’aumento del 17-OHP può essere utile nella diagnosi di CAH.
La CAH rappresenta un gruppo di patologie causate dalla mutazione di uno specifico gene e associate alla carenza degli enzimi correlati al cortisolo. Circa il 90% dei casi di CAH è dovuta a mutazioni presenti nel gene della 21-idrossilasi (noti anche come CYP-21 o P450c1 o CYP21A2) e può essere rilevata grazie all'accumulo di 17-OHP nel sangue. La malattia è manifesta nel caso in cui entrambi i geni, sia quello di origine materna che paterna, presentano una mutazione in grado di diminuire o inibire totalmente l'attività enzimatica dell'enzima codificato dal gene mutato. I genitori di pazienti affetti possono pertanto essere portatori asintomatici.
La CAH è ereditaria sia nella forma lieve che nella forma più grave.
- Nella forma più grave di CAH, i bambini possono nascere con gravi deficit di aldosterone e cortisolo, e necessitare di attenzioni mediche. Queste forme più gravi possono essere rilevate durante gli screening neonatali o anche durante l’infanzia. Se non rilevate, possono causare fin dalla prima infanzia segni e sintomi come vomito, svogliatezza, perdita di energie (letargia), inappetenza, difetti della crescita, disidratazione e abbassamenti della pressione, in particolare in caso di episodi di malessere.
Nelle bambine l’eccesso di androgeni può portare a virilizzazione, ossia allo sviluppo di caratteristiche sessuali maschili. Possono avere organi sessuali non ben definiti (genitali ambigui) con conseguente difficoltà nell’attribuzione del sesso. Possono inoltre avere una crescita eccessiva di peluria in faccia e nel corpo (irsutismo), altri segni di virilizzazione e, durante l’adolescenza, ciclo mestruale irregolare. I maschi affetti da forme gravi di CAH invece, appaiono normali alla nascita ma possono sviluppare prematuramente i caratteri sessuali maschili secondari ed avere problemi di fertilità. - Nelle più frequenti forme lievi di CAH, può esserci solo un deficit parziale della 21-idrossilasi. In queste forme, dette anche tardive o non classiche, i sintomi possono risultare evidenti sia a partire dall’infanzia, che dall’adolescenza o dall’età adulta. I sintomi possono essere di varia natura, svilupparsi lentamente e variare da persona a persona. Nonostante queste forme non siano pericolose per la vita, possono causare problemi della crescita, sviluppo e pubertà nei bambini e possono essere causa di infertilità nell’adulto.
Come e Perchè
Quali informazioni è possibile ottenere?
Il test del 17-idrossiprogesterone (17-OHP) viene utilizzato nello screening dell’iperplasia surrenale congenita (CAH) e, insieme ad altri test, può essere utilizzata come sostegno alla diagnosi ed al monitoraggio della patologia.
Screening
- La misura del 17 idrossiprogesterone (17-OHP) viene eseguita di routine come parte dello screening neonatale negli Stati Uniti e all’interno di alcun protocolli di valutazione effettuati in alcune regioni italiane.
- Il test di 17-OHP può essere usato per diagnosticare la CAH in ragazzi e adulti prima della comparsa dei sintomi, o per la conferma di una diagnosi di CAH in persone sintomatiche
Diagnosi
- La misura del 17-OHP nel sangue può essere utilizzatacome supporto alla diagnosi di CAH negli adolescenti e negli adulti con una forma lieve o ad inizio tardivo.
Monitoraggio
- In soggetti nei quali sia stata diagnosticata laCAH, la misura del 17-OHP, insieme alla misura della renina, dell’androstenedione e del testosterone, può essere utile nel monitoraggio della terapia.
Esclusione di CAH
- La misura del 17-OHP può anche essere utilizzato per escludere la CAH in donne con segni virilizzazione e/o disordini del ciclo mestruale. Ad esempio può essere richiesto in donne nelle quali vi sia un sospetto di sindrome dell’ovaio policistico, infertilità e, raramente, quando si sospetti un tumore ovarico o del surrene.
La misura del 17-OHP può produrre un numero significativo di risultati falsi positivi. Se i livelli di 17-OHP sono elevati ma non così alti da permettere una diagnosi di CAH, allora devono essere effettuati altri test, come:
- Androstenedione
- Testosterone
- Attività della renina plasmatica
Altri test
- Può essere effettuato un test di stimolo dell’ACTH come test di follow-up. In presenza di CAH, la stimolazione ACTH determina un consistente aumento di 17-OHP.
- Potrebbe essere eseguito un test genetico molecolare per rilevare le mutazioni del gene CYP21A2,causa di questa patologia.
- Per determinare il sesso del bambino e per rilevare il difetto cromosomico responsabile, è possibile effettuare un esame del cariotipo.
- La misura degli elettroliti può essere utile per valutare i livelli di sodio e potassio.
Quando viene prescritto?
Cosa significa il risultato del test?
C’è altro da sapere?
Domande Frequenti
Una persona nella quale non siano state rilevate mutazioni genetiche del gene CYP21A2 durante il test genetico, può comunque essere affetta da CAH?
Sì. Il Test rileva le mutazioni più comuni ma non rileva quelle rare. Nel caso esista una documentata familiarità per una specifica mutazione, allora verrà ricercata quella specifica mutazione. Inoltre i test genetici rilevano solo mutazioni responsabili del deficit della 21-idrossilasi. Alcune forme, più rare, di CAH possono non avere mutazioni sul gene CYP21A2.
Nel caso in cui una persona fosse affetta da CAH, deve avvisare i medici?
Nel caso in cui uno dei membri di una famiglia sia affetto da CAH, devono essere testati anche gli altri membri?
La misura del 17-OHP può essere effettuata ambulatorialmente?
Come può essere determinato il sesso di un nascituro nel caso non sia chiaramente determinabile?
Quanto tempo è necessario per l’esecuzione del test?
Pagine Correlate
In questo sito
Altrove sul web
Hormone Health Network: Congenital Adrenal Hyperplasia
The Cares Foundation: What is CAH?
The Magic Foundation: Congenital adrenal hyperplasia
National Institute of Child Health and Human Development: Congenital Adrenal Hy…
National Institutes of Health: Facts about Congenital Adrenal Hyperplasia (CAH)
Pediatric Endocrine Society Fact Sheet: Congenital Adrenal Hyperplasia (CAH)
Iperplasia Surrenalica Congenita (Sindrome Adreno Genitale) Da Deficit Di 21-Idrossilasi
Fonti
Fonti utilizzate nella revisione corrente
2017 review performed by Donald Walt Chandler PhD, Endocrine Sciences, Labcorp.
Witchel SF. Congenital Adrenal Hyperplasia. J Pediatr Adolesc Gynecol. 2017. Apr 24. pii: S1083-3188(16)30343-6. doi: 10.1016/j.jpag.2017.04.001. [Epub ahead of print] Review. PubMed PMID: 28450075.
White, PC and Speiser, PW. Congenital Adrenal Hyperplasia due to 21-Hydroxylase Deficiency. Endocrine Reviews 21(3): 245–291. PubMed PMID: 10857554.
Merke, DP and Bornstein, SR. Congenital adrenal hyperplasia. Lancet 2005; 365: 2125–36. PubMed PMID: 15964450.
Auchus, R. Management Considerations for the Adult With Congenital Adrenal Hyperplasia. Molecular and Cellular Endocrinology 408 (2015) 190–197. PMID: 25643980.
Fonti utilizzate nelle precedenti revisioni
